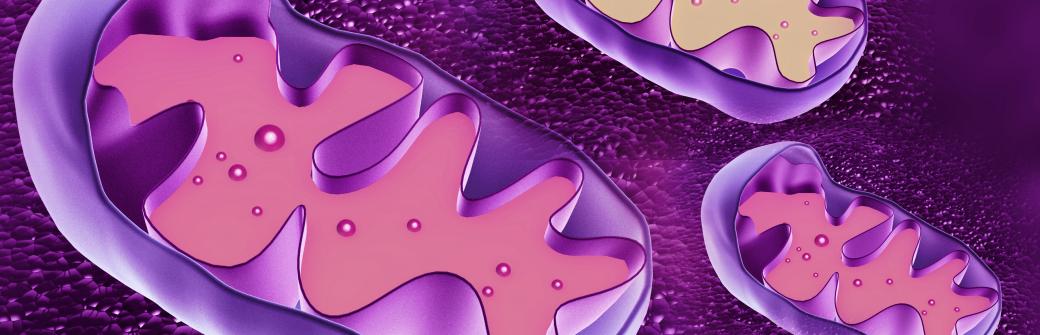
Nasze magazyny
Nasze magazyny



Chociaż mózg stanowi zaledwie ok. 2% ciężaru ciała, to nawet w stanie spoczynku zużywa 20% całkowitej energii niezbędnej dla organizmu.
Tkanki o wysokim zapotrzebowaniu energetycznym są w szczególny sposób uzależnione od mitochondriów - struktur wewnątrzkomórkowych odpowiedzialnych za większość produkcji energii w organizmie - i z tego względu mają też najniższy próg wykazywania objawów dysfunkcji mitochondrialnej. Ponieważ neurony potrzebują dużych ilości energii, by pełnić swe wyspecjalizowane funkcje, ośrodkowy układ nerwowy jest często jednym z pierwszych, które przejawiają ewidentne oznaki jej niedoborów.
Mitochondria a neurodegeneracja
Zasilacze komórek
Mitochondria to elektrownie, czy też fabryki energii komórek. Są to organella, które działają jak komórkowy układ trawienny, pobierający składniki odżywcze, rozkładający je i wytwarzający energię niezbędną do funkcjonowania komórki.
Ten proces znany jest powszechnie jako oddychanie komórkowe, a większość reakcji chemicznych zachodzących w jego ramach odbywa się właśnie w mitochondriach.
Struktury te są bardzo małe, lecz w drodze ewolucji idealnie dostosowały się do maksymalnie wydajnego wykonywania swojej ciężkiej pracy - posiadają nawet własne DNA, zwane mtDNA, kodujące wyspecjalizowane białka niezbędne w procesie oddychania komórkowego.
Każda komórka zawiera setki, a nawet tysiące mitochondriów. Szczególnie obficie występują one w sercu i mięśniach szkieletowych (wymagających dużych ilości energii do wykonywania pracy mechanicznej) oraz w większości narządów (takich jak trzustka z jej biosyntezą insuliny czy wątroba, w której odbywa się detoksykacja), a szczególnie w mózgu (gdzie komórki nerwowe wymagają ogromnych ilości energii).
Już w 1999 r. przeglądy badań naukowych zaczęły podsumowywać coraz liczniejsze dowody roli mitochondriów w procesie neurodegeneracji. Jak stwierdzili wówczas naukowcy z University of Virginia Health Sciences Center, "staje się jasne, że nawet subtelne zmiany funkcjonalne tych podstawowych komórkowych wytworników energii mogą prowadzić do zdradliwych, patologicznych zmian w naszych neuronach"1.
Autorzy przedstawili zarys teorii neurodegeneracji opartej na błędnym kole mutacji DNA, spadku energii i uszkodzeniach wywołanych przez wolne rodniki - a więc na tej samej zasadzie, jaką obecnie odnajduje się w szeregu innych schorzeń, i jaką potwierdzają dalsze badania, prowadzone przez ostatnie 20 lat.
Analizy tego typu potwierdzają rolę nieprawidłowej dynamiki mitochondriów w śmierci komórkowej neuronów i w zapoczątkowywaniu chorób Alzheimera, Parkinsona i Huntingtona. Wprawdzie wiele zaburzeń zdrowotnych, a także zmian związanych z wiekiem oraz neurodegeneracyjnych wynika ze zbliżonych przyczyn, jednak fizjologia mózgu jest pod pewnymi względami wyjątkowa, a jego stany patologiczne ujawniają interesujące mechanizmy i cechy.
Jak sygnały mózgowe zużywają energię
Sieć neuronów w całym ciele steruje myślami, ruchami i wszystkimi zmysłami, wysyłając i odbierając różnorodne neuroprzekaźniki (związki chemiczne przenoszące sygnały mózgowe) w punktach komunikacji między komórkami zwanymi synapsami. Neuroprzekaźniki uwalniane są z maleńkich wypustek ułożonych wzdłuż aksonu neuronu, zwanych kolbkami presynaptycznymi, a następnie wiążą się z receptorami na błonie sąsiedniej komórki.
Nie jest to jednak proces identyczny we wszystkich przypadkach - komórki mózgowe komunikują się ze sobą nawzajem ze zróżnicowaną siłą i częstotliwością. Czasem mówią głośno i wyraźnie, a kiedy indziej szepczą lub mamroczą.
Przez wiele lat badacze stawiali pytania, dlaczego i w jaki sposób neurony tak często zmieniają intensywność swojej pracy. Badanie przeprowadzone przez naukowców z amerykańskich Narodowych Instytutów Zdrowia pokazało, że gwałtownie poruszające się mitochondria emitują dosłownie wybuchy energii, które, jak się wydaje, regulują komunikację neuronalną1. Kolbki presynaptyczne pomagają sterować siłą wysyłanych sygnałów poprzez regulowanie ilości uwalnianych neuroprzekaźników, jak również sposobu ich uwalniania. Produkcja neuroprzekaźników, ich pakowanie i uwalnianie, a także odbieranie lub usuwanie to procesy wymagające energii.
Wcześniejsze badania wykazały, że mitochondria mogą szybko poruszać się wzdłuż aksonów, jak gdyby tańcząc od jednej kolbki do drugiej.
Badacze z NIH poprowadzili tę obserwację o krok dalej, wykazując, że przemieszczające się mitochondria mogą sterować siłą sygnałów wysyłanych z kolbek. Wykorzystali oni zaawansowane techniki, by obserwować, jak mitochondria poruszają się między nimi podczas uwalniania neuroprzekaźników, i odkryli, że kolbki wysyłają sygnały o niezmiennie dużej sile tylko wtedy, gdy w pobliżu znajdują się mitochondria. Jeśli ich nie ma, siła wysyłanych sygnałów podlega wahaniom.
Te rezultaty sugerują, że obecność nieruchomych mitochondriów przy synapsach poprawia stabilność i siłę sygnałów nerwowych. Wcześniej wykazano, że porusza się ok. 1/3 wszystkich mitochondriów w aksonach, podczas gdy reszta pozostaje nieruchoma2.
Komunikacja komórek nerwowych jest najwyraźniej ściśle sterowana przez bardzo dynamiczne zdarzenia zachodzące w licznych synapsach. Odkrycie to będzie bardzo cenne dla zrozumienia, w jaki sposób mitochondria uczestniczą nie tylko w chorobach neurodegeneracyjnych, ale także we wszelkich schorzeniach neurologicznych, w których występują zmiany sygnalizacji komórek nerwowych, jak np. depresja lub ADHD.
Bibliografia
- Cell Rep, 2013; 4: 413–9
- Cell, 2008; 132: 137–48
Wrażliwy umysł
Mózg jest szczególnie czuły na uszkodzenia wywoływane przez wolne rodniki (ze względu na jego duże zapotrzebowanie na tlen oraz wysoką zawartość tłuszczu). Logiczne wydaje się więc założenie, że jego układ obrony antyoksydacyjnej powinien być szczególnie skuteczny.
Niestety tak nie jest - ten delikatny narząd jest stosunkowo słabo chroniony przed uszkodzeniami powodowanymi przez wolne rodniki. W wyniku tego w komórkach mózgu z upływem czasu stopniowo nawarstwiają się uszkodzenia oksydacyjne. Dotyczy to każdego z nas, lecz szczególnie niepokojące jest u osób z genetyczną lub środowiskową predyspozycją do degeneracji neurologicznej.
Większość zawartego w mózgu tłuszczu znajduje się w błonach komórkowych, długich "ramionach" i "gałęziach" (zwanych aksonami i dendrytami), wychodzących z komórek, a także w ich mitochondriach. W miarę jak się starzejemy, coraz więcej lipidów ulega utlenianiu pod wpływem wysokiego poziomu tlenu oraz wolnych rodników, a podatność mózgu na choroby degeneracyjne wzrasta.
Zachowanie zdrowia mitochondriów jest ważną strategią w zapobieganiu powolnemu spadkowi naszych zdolności umysłowych w miarę upływu czasu. Już pod koniec lat 80. naukowcy z amerykańskich Narodowych Instytutów Zdrowia (NIH) zasugerowali, iż ekscytotoksyczność (toksyczność w wyniku nadmiernej stymulacji komórek nerwowych) pojawia się wtedy, gdy spada poziom energii neuronów2. W normalnej sytuacji neuroprzekaźnik glutaminian sprawnie przekazuje impulsy pobudzające z jednego neuronu do drugiego.
W stanie neurodegeneracji natomiast mózg nabiera chronicznej nadwrażliwości na glutaminian, który staje się wtedy dla komórek mózgowych powoli działającą toksyną. Dla mitochondriów oznacza to, że stale zmuszane są do produkowania większej ilości energii - większej niż rzeczywiste zapotrzebowanie neuronów. Wraz ze wzmożonym tempem aktywności wzrasta produkcja wolnych rodników, co z czasem prowadzi do przyspieszonego niszczenia mitochondriów. Ten łańcuch zdarzeń powoduje w końcu dysfunkcję neuronów.
Badacze z NIH, opierając się na swym odkryciu, iż obecność mitochondriów w pobliżu synapsy wpływa na siłę sygnałów neuronu (patrz ramka), manipulowali ich ruchem za pomocą zmian poziomu syntafiliny - białka pomagającego w kotwiczeniu mitochondriów wewnątrz aksonów. Usuwanie syntafiliny sprawiało, że te małe elektrownie poruszały się szybciej, a zapis elektryczny neuronów wykazywał wielkie fluktuacje wysyłanych przez nie sygnałów. I odwrotnie - podwyższenie poziomu białka spowalniało ruch mitochondriów i skutkowało wysyłaniem przez kolbki synaptyczne sygnałów o stałej sile.
Naukowcy odkryli też, że blokowanie produkcji ATP - związku odpowiedzialnego za magazynowanie energii komórkowej - obniżało siłę wysyłanych sygnałów, nawet jeśli mitochondria znajdowały się w pobliżu kolbek synaptycznych. Jak stwierdzono, problemy z produkcją energii i ruchem mitochondriów w neuronach odgrywają rolę w chorobie Alzheimera, Parkinsona, stwardnieniu zanikowym bocznym (ALS) i w innych poważnych schorzeniach neurodegeneracyjnych. Wspomniane badanie dostarczyło kluczowego elementu układanki i dało nam jeszcze więcej powodów, by w ich badaniu koncentrować się na mitochondriach i energii komórkowej3.

|
Jak degeneruje się mózg
Na poziomie komórkowym narząd dotknięty chorobą Alzheimera wykazuje rozległy zanik neuronów i wysoki poziom nierozpuszczalnych włóknistych złogów, znanych jako płytki starcze i splątki neurofibrylarne. Rdzeniem płytek jest toksyczne białko o nazwie amyloid beta - znak rozpoznawczy choroby - atakujące komórki na kilku frontach. Generuje ono wolne rodniki, uszkadza mtDNA, osłabia bioenergetykę komórkową i zmienia fałdowanie innych białek, w wyniku czego tworzą one następnie splątki neurofibrylarne.
Istnieją jednak dowody sugerujące, że wytwarzanie amyloidu beta to sposób, w jaki mózg broni się przed stresem oksydacyjnym. Innymi słowy jego powstawanie jest konsekwencją choroby Alzheimera, a nie jej przyczyną.
Zgodnie z rezultatami niektórych analiz stopień niesprawności w chorobie Alzheimera skorelowany jest z poziomem bioenergetycznego upośledzenia mózgu. Niedawny przegląd sugeruje wręcz, że komórkowa produkcja energii może być tu lepszym wskaźnikiem niż płytki starcze4.
W badaniu przeprowadzonym przez ośrodek noszący obecnie nazwę Burke Neurological Institute stopień klinicznej niesprawności pacjenta nie miał związku z gęstością płytek starczych, był za to skorelowany z nieprawidłowościami mitochondriów, odgrywającymi rolę w energetyce komórkowej5. Ważnym źródłem stresu oksydacyjnego w mózgu jest silny wolny rodnik - nadtlenoazotyn (powstający z tlenku azotu), który utlenia lipidy w błonach komórek nerwowych. Powoduje to wytwarzanie wysoce toksycznego produktu ubocznego o nazwie hydroksynonenal (HNE), którego nadmierny poziom stwierdza się w wielu regionach mózgu pacjentów cierpiących na chorobę Alzheimera. HNE zabija komórki mózgowe nie tylko w sposób bezpośredni, ale także pośrednio, czyniąc je bardziej podatnymi na ekscytotoksyczność.
Dotychczasowe badania nie zdołały ustalić pojedynczej przyczyny leżącej u podłoża choroby Alzheimera, ale w 1997 r. interesującą teorię wieloczynnikową zaproponował Wan-Tao Ying z University of New Mexico6. W myśl tej hipotezy choroba Alzheimera rozwija się w wyniku wzajemnych oddziaływań 4 przyczyn: nierównowagi APP (białka prekursorowego amyloidu) i wapnia, uszkodzeń wywołanych przez wolne rodniki oraz niedoboru energii. Ying cytuje w swej pracy badania pokazujące, że każdy z tych czynników zarówno wzmacnia, jak i jest wzmacniany przez 3 pozostałe.
Mózg i choroba Parkinsona: czy tylko lewodopa?
Nowe doniesienia pokazują, że dysfunkcja mitochondrialna i zaburzenia komórkowej produkcji energii odgrywają istotną rolę w postępie choroby Parkinsona. Badania prowadzone na zwierzęcych modelach sugerują, że koenzym Q10 (CoQ10), przeciwutleniacz odgrywający ważną rolę w funkcjonowaniu mitochondriów, może chronić komórki mózgowe przed neurotoksycznością i ekscytotoksycznością nawet w tych przypadkach, w których inne silne antyoksydanty są bezradne.
W chorobie Parkinsona śmierć komórek następuje przede wszystkim wśród neuronów istoty czarnej - części mózgu koordynującej ruchy. Neurony te wytwarzają neuroprzekaźnik - dopaminę. Ich śmierć obniża jej poziom, prowadząc w końcu do sztywności mięśni, drżenia i trudności z zapoczątkowaniem ruchu.
W badaniach stwierdzono, że istota czarna to część mózgu wykazująca największą ilość mutacji mtDNA, a mitochondria u pacjentów z chorobą Parkinsona przejawiają pewne niedomagania7.
Inne badania, prowadzone na szczurach, wykazały zależny od dawki wzrost poziomu wolnych rodników w mitochondriach, gdy zwierzętom podawano lewodopę (L-dopa), prekursor dopaminy, stanowiący podstawową terapię farmakologiczną u pacjentów z chorobą Parkinsona8. Badania te były pierwszym sygnałem, iż zwiększanie ilości substancji występującej - naszym zdaniem - w niedoborze może nie być najlepszym rozwiązaniem problemu.
Badania nad dysfunkcją mitochondrialną w parkinsonie zrodziły poważne pytania w odniesieniu do konwencjonalnego stosowania lewodopy w leczeniu tej choroby. Jest ona przepisywana, ponieważ łagodzi objawy (przynajmniej czasowo), nie likwiduje jednak stanu patologicznego leżącego u źródeł choroby. Tymczasem coraz więcej dowodów wskazuje na to, że lewodopa może wręcz nasilać niektóre z przyczyn wywołujących parkinsona. Jak przecież dobrze wiadomo, terapia z jej użyciem w końcu traci swą skuteczność, a objawy powracają z większą intensywnością niż wcześniej. Skoro tak, być może nadszedł czas, by na nowo rozważyć koszty i korzyści wynikające ze stosowania tego leku.
Mitochondria pacjentów z chorobą Parkinsona wykazują również pewne zahamowanie aktywności (choć stosunkowo łagodniejsze niż to, które stwierdza się w chorobie Alzheimera), a także względny niedobór kompleksu dehydrogenazy alfa-ketoglutaranu (KGDHC), głównego enzymu występującego w macierzy mitochondrialnej. KGDHC produkuje dinukleotyd nikotynoamidoadeninowy (NADH) - związek transportujący elektrony, niezbędny w procesie oddychania komórkowego, którego zasoby są znacząco zubożone w pewnych obszarach mózgu pacjentów z chorobą Parkinsona9.
Co ciekawe, obniżenie poziomu KGDHC stwierdzano również w korze mózgowej pacjentów z alzheimerem.
M. Flint Beal, wybitny neurolog i profesor neurobiologii w Cornell University, poświęcił wiele lat na udowodnienie, że koenzym Q10 ma właściwości neuroprotekcyjne, które mogą okazać się pomocne w takich chorobach jak parkinson i huntington, a jego hipotezę potwierdza coraz więcej dowodów naukowych.
Nowy obrońca - PQQ
Choć to koenzym Q10 jest bezsprzecznie najważniejszym sprzymierzeńcem mitochondriów, na początku 2010 r. badacze odkryli, że również pirolochinolinochinon (PQQ) sprzyja poprawie ich pracy. Nie tylko chroni on mitochondria przed uszkodzeniami oksydacyjnymi, lecz także stymuluje wzrost nowych organelli1.
Związkowi temu przypisano szereg właściwości fizjologicznych, m.in. ochronę komórek nerwowych, sprzyjanie wzrostowi nerwów oraz biogenezę mitochondriów.
Mocne dowody wskazują na to, że PQQ może odgrywać istotną rolę w ścieżkach ważnych dla sygnalizacji komórkowej. Badania na zwierzętach pokazały, że chroni on komórki nerwowe przed degeneracją i uszkodzeniami, a nawet sprzyja ich wzrostowi i pomaga w tworzeniu nowych synaps między neuronami, co ma decydujące znaczenie dla pamięci. Obecnie wstępne badania prowadzone na ludziach zaczynają potwierdzać potencjalne korzyści zdrowotne PQQ.
Badania sugerują także, iż związek ten może mieć działanie przeciwzapalne, być skutecznym neuroprotektorem (zmniejszającym uszkodzenia mózgu podczas udaru i chroniącym przez nadmierną stymulacją ekscytotoksyczną), jak również stymulatorem czynnika wzrostu nerwów - głównego białka odgrywającego rolę we wzroście i przeżyciu komórek nerwowych2.
Sugerowana dawka dzienna: W jednym z badań 20 mg PQQ przyjmowane doustnie każdego dnia poprawiło pamięć krótkotrwałą, uwagę, koncentrację oraz zdolność identyfikowania i przetwarzania informacji u zdrowych osób dorosłych. Efekty te znacznie nasiliły się, gdy dodano suplementację koenzymem Q10.
Bibliografia
- J Biol Chem, 2010; 285: 142-52
- Biosci Biotechnol Biochem, 2016; 80: 13-22
Zespół Beala wykazał, że podawanie koenzymu Q10 szczurom w średnim i podeszłym wieku pozwalało przywrócić stężenie tego składnika odżywczego właściwe dla młodszych osobników. Poziom Q10 wzrastał o 10-40% w mitochondriach tego obszaru kory mózgowej, w którym powstaje większość wyższych myśli10.
W późniejszym badaniu na myszach doustna suplementacja koenzymem Q10 spowalniała toksyczny wpływ trucizny znanej z wywoływania u zwierząt zespołu parkinsonowskiego. Po kilku tygodniach narażenia na działanie tej substancji u wszystkich zwierząt spadły stężenie dopaminy i gęstość aksonów dopaminergicznych w obszarach otaczających istotę czarną, ale u myszy, którym wcześniej podawano koenzym Q10, poziomy te były znacznie wyższe (odpowiednio: 37 i 62%), co potwierdziło, iż deficyt bioenergetyczny jest jednym z komponentów choroby Parkinsona11.
Wspomagacze mózgu
CoQ10 Koenzym Q10, zbliżona do witamin cząsteczka występująca naturalnie w niemal każdej komórce naszego ciała, jest przeciwutleniaczem, stabilizatorem błon komórkowych oraz bardzo ważnym elementem procesu produkcji energii w mitochondriach. Ma również działanie neuroprotekcyjne.
Badania wykazały, że koenzym Q10 może zwiększać liczbę mitochondriów w mózgu i chronić zwierzęta przed neurodegeneracją wywoływaną zarówno przez toksyny, jak i mutacje genów12. Wprawdzie przyjmujemy niewielkie ilości koenzymu Q10 z pożywieniem, jest to jednak zaledwie kilka miligramów dziennie - o wiele za mało, by organizm mógł odnieść z tego korzyści - a w miarę, jak przybywa nam lat, suplementacja staje się coraz bardziej istotna.
Naukowcy opracowują nowe systemy dostarczania koenzymu Q10, mające podnieść jego skuteczność13. Preparaty na bazie oleju (zazwyczaj w kapsułkach żelowych) są, jak się uważa, lepiej przyswajalne przez organizm, podobnie jak formy liposomalne. Wydaje się, że ubichinol (zredukowany koenzym Q10) jest znacznie lepiej wchłaniany niż ubichinon (utleniony koenzym Q10), a jeszcze większą absorpcję wykazuje ubichinol rozpuszczalny w wodzie (solubilizowany).
Sugerowana dawka dzienna: w schorzeniach neurologicznych, 600-3000 mg, przyjmowane w mniejszych dawkach wraz z posiłkami w ciągu całego dnia.
D-ryboza Ten pięciowęglowy cukier prosty odgrywa ważną rolę w syntezie energetycznej (jako strukturalny komponent ATP odpowiada za magazynowanie energii). W latach 80. badacze odkryli, że przy suplementacji D-rybozą, podawaną przed lub tuż po niedokrwieniu serca, mięsień sercowy wykazujący niedobór energetyczny był w stanie odzyskać wcześniejszy poziom energii komórkowej14.
Sugerowana dawka dzienna: 3-5 g.
L-karnityna Lewokarnityna, związek chemiczny występujący naturalnie u wszystkich gatunków zwierząt, transportuje do mitochondriów tłuszcze do produkcji ATP.
Przyjmowanie L-karnityny w diecie odbywa się przede wszystkim przez spożycie produktów pochodzenia zwierzęcego, takich jak czerwone mięso, drób, ryby i nabiał, podczas gdy w produktach roślinnych związek ten występuje tylko w nieznacznych ilościach.
Standardowa dieta dostarcza 6-15 milimoli na kilogram dziennie, natomiast wegetariańska - mniej niż 1 milimol na kilogram dziennie.
By L-karnityna mogła być prawidłowo syntetyzowana, niezbędne są też inne składniki odżywcze, takie jak żelazo, witamina C, tlen, 5-fosforan pirydoksalu (biologicznie aktywna forma witaminy B6) oraz witamina B3.
Sugerowana dawka dzienna: 500-2000 mg.
Magnez Ten pierwiastek ma decydujące znaczenie jako kofaktor ponad 300 reakcji biochemicznych w organizmie, włącznie z produkcją ATP.
Mitochondria działają jak wewnątrzkomórkowe magazyny magnezu, a znaczna część tego pierwiastka w naszym organizmie występuje w postaci związanej z ATP, co pomaga stabilizować cząsteczki i umożliwia organizmowi korzystanie z nich.
Sugerowana dawka dzienna: 400-800 mg.
Kwas alfa-liponowy ALA, przeciwutleniacz występujący w mitochondriach, może być w optymalnych warunkach produkowany przez organizm na potrzeby jego funkcji metabolicznych, lecz dostarczany jako suplement może działać jako przeciwutleniacz rozpuszczalny zarówno w wodzie, jak i w tłuszczach.
Organizm wykorzystuje tylko jedną jego postać, zwaną R(+), która sprzedawana jest w formie chłodzonej (i powinna być przechowywana w domu w lodówce). Należy unikać wystawiania ALA na działanie wszelkich źródeł ciepła.
Sugerowana dawka dzienna: 300-600 mg.
Kreatyna Ludzki organizm wytwarza ją z aminokwasów: metioniny, glicyny i argininy. Nasze ciało zawiera przeciętnie ok. 120 g kreatyny zmagazynowanej w postaci fosforanu (znanego również jako fosfokreatyna). Niektóre produkty spożywcze (takie jak wołowina i ryby) odznaczają się stosunkowo wysoką zawartością tego związku.
Rosnąca liczba dowodów potwierdza, że kreatyna może chronić mózg przed czynnikami neurotoksycznymi i niektórymi rodzajami uszkodzeń. Badania wykazały również, że ma wysoką skuteczność neuroprotekcyjną wobec różnorodnych czynników neurotoksycznych15.
Sugerowana dawka dzienna: 2-25 g, zależnie od wagi ciała (0,1g/kg).
Resweratrol i pterostylben Choć druga nazwa może brzmieć nieznajomo, tak naprawdę te substancje są bardzo podobne i wykazują równie dobre efekty. Naukowcy odkryli wręcz, że pterostylben to bliski krewny resweratrolu - związku występującego w czerwonych winogronach, który zyskał sobie popularność ze względu na korzystne działanie dla zdrowia mózgu. Występuje przede wszystkim w czarnych jagodach, a także w winogronach i korze indyjskiego drzewa kino (stosowanego od stuleci w tradycyjnej medycynie ajurwedyjskiej). Jak się wydaje, oddziałuje on na mózg synergistycznie z resweratrolem16.
Pterostylben wywołuje korzystne zmiany, takie jak podwyższenie poziomu specyficznych białek mózgowych związanych z poprawą pamięci. Wiele z tych korzyści ma oczywiście związek z naszymi skromnymi mitochondriami.
Sugerowana dawka dzienna: 150-500 mg resweratrolu, 100-500 mg pterostylbenu.

Artykuł ukazał się pt. "Jak doładować szare komórki" w numerze Październik 2018 >>
Bibliografia
- Brain Res Brain Res Rev, 1999; 29: 1-25
- Brain Res,1988; 451: 205-12
- Cell Rep, 2013; 4: 413-9
- Curr Pharm Des, 2013; 19: 6440-50
- J Mol Neurosci, 2001; 16: 41-8
- Gerontology, 1997; 43: 242-53
- Nat Commun, 2016; 7: 13548
- Neuroreport, 1994; 5: 1009-11
- Ann Neurol, 1994; 35: 204-10
- Ann Neurol, 1994; 36: 882-8
- Brain Res, 1998; 783: 109-14
- Proc Natl Acad Sci U S A, 1998; 95: 8892-7
- Curr Drug Deliv, 2016; 13: 1184-1204
- J Thorac Cardiovasc Surg, 1982; 83: 390-8
- Amino Acids, 2011; 40: 1305-13
- Neurochem Int, 2015; 89: 227-33